国际医疗器械监管机构论坛(IMDRF,原名GHTF)在2011年完成了对UDI实施原则的协调,提出了《医疗器械UDI系统》指导性最终文件,推动UDI作为全球医疗器械上市后追溯的基本手段,并推荐UDI采用开放的GS1国际物品编码标准。欧洲医疗器械行业协会(Eucomed)在此框架下进一步制定了《基于风险管理的不同包装级别医疗器械UDI标识要求》。
美国等先进国家也积极制定UDI政策法规,推动UDI在本国的规范管理与应用实施。
2014年9月24日开始
-所有FDAClassIII第三类医疗器械;FDAPHSAct法案管制的器械,都必须在器械的标签和包装上标注FDAUDI信息,日期格式必须符合21CFR801。18的规定,并及时报送FDAGUDID数据库;
-FDAClassIIIStand-aloneSoftware被FDA作为第三类别管制的独立软件必须提供UDI信息
2015年9月25日开始
-Implantable,life-supporting,andlife-sustainingdevices植入式医疗器械,用于生命支持和维持的医疗器械的标签和包装都必须标注UDI信息,日期格式必须符合21CFR801。18的规定;
-如果上述器械为重复使用器械并在使用前可被再处理,那么必须进行永久性UDI标识;
-用于生命支持或维持器械的独立软件必须标识UDI;
-上述器械UDI数据必须报送FDAGUDID数据库;
2016年9月24日开始
-UDI管制的ClassIII可重复使用并且在使用前可以被再处理的医疗器械都需要进行永久性UDI标识;
-FDAClassII二类器械的产品标签和包装必须标识UDI,日期格式必须符合21CFR801。18的规定;FDAClassIIStand-aloneSoftware被FDA作为第二类别管制的独立软件必须提供UDI信息;
-FDAClassII二类器械的UDI数据以及产品关键数据必须报送FDAGUDID数据库;
2018年9月24日开始
-FDAClassII可重复使用的并且使用前可以被再处理的FDA管制的二类器械必须进行永久性UDI标识;
-FDAClassI一类医疗器械和未被划分级别ClassI,II或III的器械都必须标识UDI;所有这些器械,包含豁免UDI的器械,日期标注都必须符合UDI法规;
-上述器械的UDI信息必须及时报送FDAGUDID数据库;
-FDAClassI一类的Stand-alonesoftware独立软件必须提供UDI;
2020年9月24日开始
-所有FDAClassI一类器械和未被分类为ClassI,II,III的器械,如果可以重复使用或使用前可以被再处理,都必须在产品上进行永久性UDI标注。
中国于1991年4月由中国物品编码中心代表,加入国际物品编码协会(GS1),负责推广国际通用的、开放的、跨行业的全球统一编码标识系统和供应链管理标准,向社会提供公共服务平台和标准化解决方案。
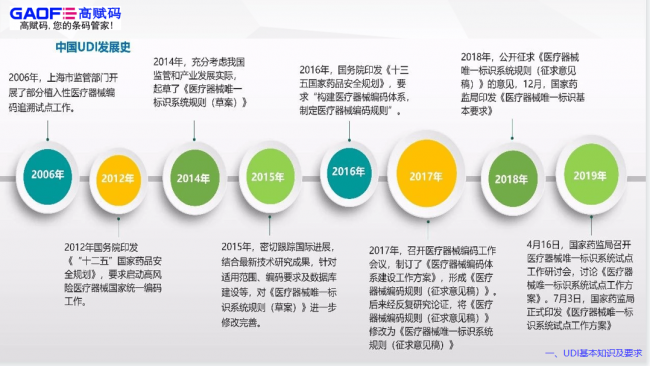
2018年02月27日发布国家总局办公厅公开征求医疗器械唯一标识系统规则(征求意见稿)意见
2019年07月03日发布国家药监局综合司国家卫生健康委办公厅关于印发医疗器械唯一标识系统试点工作方案的通知(药监综械注〔2019〕56号)